Pedigree Reconstruction
sequoia.RdPerform pedigree reconstruction based on SNP data, including parentage assignment and sibship clustering.
Usage
sequoia(
GenoM = NULL,
LifeHistData = NULL,
SeqList = NULL,
Module = "ped",
Err = 1e-04,
Tfilter = -2,
Tassign = 0.5,
MaxSibshipSize = 100,
DummyPrefix = c("F", "M"),
Complex = "full",
Herm = "no",
UseAge = "yes",
args.AP = list(Flatten = NULL, Smooth = TRUE),
mtSame = NULL,
CalcLLR = FALSE,
quiet = FALSE,
Plot = NULL,
StrictGenoCheck = TRUE,
ErrFlavour = "version2.9"
)Arguments
- GenoM
numeric matrix with genotype data: One row per individual, one column per SNP, coded as 0, 1, 2, missing values as a negative number or NA. Row names must be individual IDs, column names are ignored. You can reformat data with
GenoConvert, or use other packages to get it into a genlight object and then useas.matrix.- LifeHistData
data.frame with up to 6 columns:
- ID
max. 30 characters long
- Sex
1 = female, 2 = male, 3 = unknown, 4 = hermaphrodite, other numbers or NA = unknown
- BirthYear
birth or hatching year, integer, with missing values as NA or any negative number.
- BY.min
minimum birth year, only used if BirthYear is missing
- BY.max
maximum birth year, only used if BirthYear is missing
- Year.last
Last year in which individual could have had offspring. Can e.g. in mammals be the year before death for females, and year after death for males.
"Birth year" may be in any arbitrary discrete time unit relevant to the species (day, month, decade), as long as parents are never born in the same time unit as their offspring, and only integers are used. Individuals do not need to be in the same order as in `GenoM', nor do all genotyped individuals need to be included.
- SeqList
list with output from a previous run, to be re-used in the current run. Used are elements `PedigreePar', `LifeHist', `AgePriors', `Specs', and `ErrM', and these override the corresponding input parameters. Not all of these elements need to be present, and all other elements are ignored. If
SeqList$Specsis provided, all input parameters with the same name as its items are ignored, exceptModule.- Module
one of
- pre
Only input check, return
SeqList$Specs- dup
Also check for duplicate genotypes
- par
Also perform parentage assignment (genotyped parents to genotyped offspring)
- ped
(Also) perform full pedigree reconstruction, including sibship clustering and grandparent assignment. By far the most time consuming, and may take several hours for large datasets.
- Err
assumed per-locus genotyping error rate, as a single number, or a length 3 vector with P(hom|hom), P(het|hom), P(hom|het), or a 3x3 matrix. See details below. The error rate is presumed constant across SNPs, and missingness is presumed random with respect to actual genotype. Using
Err>5% is not recommended, andErr>10% strongly discouraged. SeeErr_RADseqto convert per-allele rates at homozygous and heterozygous sites to the required length-3 vector, andErrToMfor further genotyping error details.- Tfilter
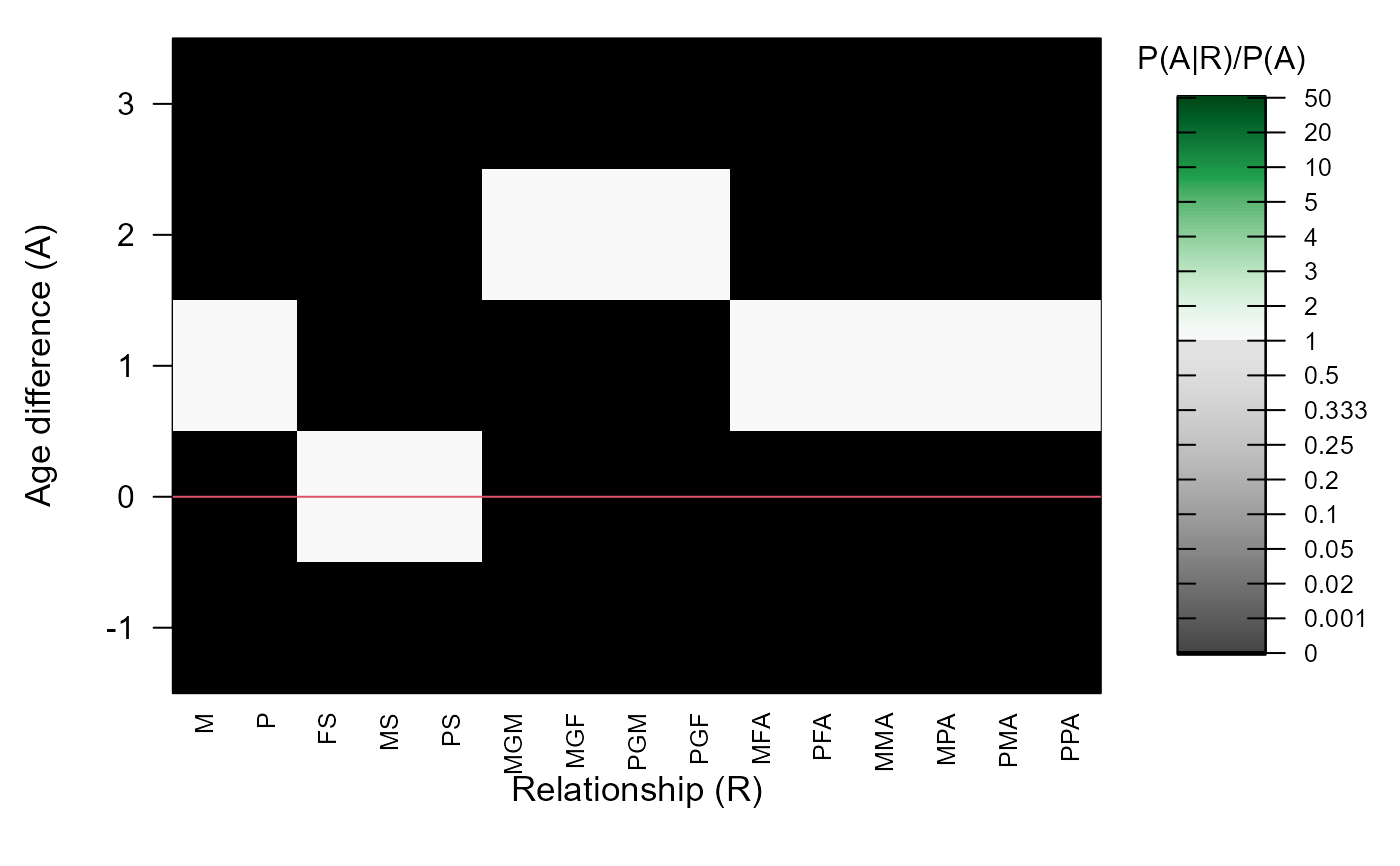
threshold log10-likelihood ratio (LLR) between a proposed relationship versus unrelated, to select candidate relatives. Typically a negative value, related to the fact that unconditional likelihoods are calculated during the filtering steps. More negative values may decrease non-assignment, but will increase computational time.
- Tassign
minimum LLR required for acceptance of proposed relationship, relative to next most likely relationship. Higher values result in more conservative assignments. Must be zero or positive.
- MaxSibshipSize
maximum number of offspring for a single individual (a generous safety margin is advised).
- DummyPrefix
character vector of length 2 with prefixes for dummy dams (mothers) and sires (fathers); maximum 20 characters each. Length 3 vector in case of hermaphrodites (or default prefix 'H').
- Complex
Breeding system complexity. Either "full" (default), "simp" (simplified, no explicit consideration of inbred relationships), "mono" (monogamous).
- Herm
Hermaphrodites, either "no", "A" (distinguish between dam and sire role, default if at least 1 individual with sex=4), or "B" (no distinction between dam and sire role). Both of the latter deal with selfing.
- UseAge
either "yes" (default), "no" (only use age differences for filtering), or "extra" (additional rounds with extra reliance on ageprior, may boost assignments but increased risk of erroneous assignments). Used during full reconstruction only.
- args.AP
list with arguments to be passed on to
MakeAgePrior, e.g. `Discrete` (non-overlapping generations), `MinAgeParent`, `MaxAgeParent`.- mtSame
matrix indicating whether individuals (might) have the same mitochondrial haplotype (1), and may thus be matrilineal relatives, or not (0). This potentially be useful to distinguish between maternal and paternal half siblings when few parents are genotyped, or between maternal and paternal parents when their sex(role) is unknown. Row names and column names should match IDs in `GenoM`. Not all individuals need to be included and order is not important. For details see the mtDNA vignette.
- CalcLLR
TRUE/FALSE; calculate log-likelihood ratios for all assigned parents (genotyped + dummy; parent vs. otherwise related). Time-consuming in large datasets. Can be done separately with
CalcOHLLR.- quiet
suppress messages: TRUE/FALSE/"verbose".
- Plot
display plots from
SnpStats, MakeAgePrior, andSummarySeq. Defaults (NULL) to TRUE when quiet=FALSE or "verbose", and FALSE when quiet=TRUE. If you get error 'figure margins too large', enlarge the plotting area (drag with mouse). Error 'invalid graphics state' can be dealt with by clearing the plotting area with dev.off().- StrictGenoCheck
Automatically exclude any individuals genotyped for <5 the unavoidable default up to version 2.4.1. Otherwise only excluded are (very nearly) monomorphic SNPs, SNPs scored for fewer than 2 individuals, and individuals scored for fewer than 2 SNPs.
- ErrFlavour
function that takes
Err(single number) as input, and returns a length 3 vector or 3x3 matrix, or choose from inbuilt options 'version2.9', 'version2.0', 'version1.3', or 'version1.1', referring to the sequoia version in which they were the default. Ignored ifErris a vector or matrix.
Value
A list with some or all of the following components, depending on
Module. All input except GenoM is included in the output.
- AgePriors
Matrix with age-difference based probability ratios for each relationship, used for full pedigree reconstruction; see
MakeAgePriorfor details. When running only parentage assignment (Module="par") the returned AgePriors has been updated to incorporate the information of the assigned parents, and is ready for use during full pedigree reconstruction.- args.AP
(input) arguments used to specify age prior matrix. If a custom ageprior was provided via
SeqList$AgePrior, this matrix is returned instead- DummyIDs
Dataframe with pedigree for dummy individuals, as well as their sex, estimated birth year (point estimate, upper and lower bound of 95% confidence interval; see also
CalcBYprobs), number of offspring, and offspring IDs. From version 2.1 onwards, this includes dummy offspring.- DupGenotype
Dataframe, duplicated genotypes (with different IDs, duplicate IDs are not allowed). The specified number of maximum mismatches is used here too. Note that this dataframe may include pairs of closely related individuals, and monozygotic twins.
- DupLifeHistID
Dataframe, row numbers of duplicated IDs in life history dataframe. For convenience only, but may signal a problem. The first entry is used.
- ErrM
(input) Error matrix; probability of observed genotype (columns) conditional on actual genotype (rows)
- ExcludedInd
Individuals in GenoM which were excluded because of a too low genotyping success rate (<50%).
- ExcludedSNPs
Column numbers of SNPs in GenoM which were excluded because of a too low genotyping success rate (<10%).
- LifeHist
(input) Dataframe with sex and birth year data. All missing birth years are coded as '-999', all missing sex as '3'.
- LifeHistPar
LifeHist with additional columns 'Sexx' (inferred Sex when assigned as part of parent-pair), 'BY.est' (mode of birth year probability distribution), 'BY.lo' (lower limit of 95% highest density region), 'BY.hi' (higher limit), inferred after parentage assignment. 'BY.est' is NA when the probability distribution is flat between 'BY.lo' and 'BY.hi'.
- LifeHistSib
as LifeHistPar, but estimated after full pedigree reconstruction
- NoLH
Vector, IDs in genotype data for which no life history data is provided.
- Pedigree
Dataframe with assigned genotyped and dummy parents from Sibship step; entries for dummy individuals are added at the bottom.
- PedigreePar
Dataframe with assigned parents from Parentage step.
- Specs
Named vector with parameter values. This includes the maximum OH for potential (parent-)parent-offspring pairs (trios), which is calculated by
CalcMaxMismatch- TotLikParents
Numeric vector, Total likelihood of the genotype data at initiation and after each iteration during Parentage.
- TotLikSib
Numeric vector, Total likelihood of the genotype data at initiation and after each iteration during Sibship clustering.
- AgePriorExtra
As AgePriors, but including columns for grandparents and avuncular pairs. NOT updated after parentage assignment, but returned as used during the run.
- DummyClones
Hermaphrodites only: female-male dummy ID pairs that refer to the same non-genotyped individual
List elements PedigreePar and Pedigree both have the following columns:
- id
Individual ID
- dam
Assigned mother, or NA
- sire
Assigned father, or NA
- LLRdam
Log10-Likelihood Ratio (LLR) of this female being the mother, versus the next most likely relationship between the focal individual and this female. See Details below for relationships considered, and see
CalcPairLLfor underlying likelihood values and further details)- LLRsire
idem, for male parent
- LLRpair
LLR for the parental pair, versus the next most likely configuration between the three individuals (with one or neither parent assigned)
- OHdam
Number of loci at which the offspring and mother are opposite homozygotes
- OHsire
idem, for father
- MEpair
Number of Mendelian errors between the offspring and the parent pair, includes OH as well as e.g. parents being opposing homozygotes, but the offspring not being a heterozygote. The offspring being OH with both parents is counted as 2 errors.
Details
For each pair of candidate relatives, the likelihoods are calculated of them being parent-offspring (PO), full siblings (FS), half siblings (HS), grandparent-grandoffspring (GG), full avuncular (niece/nephew - aunt/uncle; FA), half avuncular/great-grandparental/cousins (HA), or unrelated (U). Assignments are made if the likelihood ratio (LLR) between the focal relationship and the most likely alternative exceed the threshold Tassign.
Dummy parents of sibships are denoted by F0001, F0002, ... (mothers) and M0001, M0002, ... (fathers), are appended to the bottom of the pedigree, and may have been assigned real or dummy parents themselves (i.e. sibship-grandparents). A dummy parent is not assigned to singletons.
Full explanation of the various options and interpretation of the output is provided in the vignettes and on the package website, https://jiscah.github.io/index.html .
Genotyping error rate
The genotyping error rate Err can be specified three different ways:
A single number, which is combined with
ErrFlavourbyErrToMto create a length 3 vector (next item). By default (ErrFlavour= 'version2.9'), P(hom|hom)=$(E/2)^2$, P(het|hom)=$E-(E/2)^2$, P(hom|het)=$E/2$.a length 3 vector (NEW from version 2.6), with the probabilities to observe a actual homozygote as the other homozygote (hom|hom), to observe a homozygote as heterozygote (het|hom), and to observe an actual heterozygote as homozygote (hom|het). This assumes that the two alleles are equivalent with respect to genotyping errors, i.e. $P(AA|aa) = P(aa|AA)$, $P(aa|Aa)=P(AA|Aa)$, and $P(aA|aa)=P(aA|AA)$.
a 3x3 matrix, with the probabilities of observed genotype (columns) conditional on actual genotype (rows). Only needed when the assumption in the previous item does not hold. See
ErrToMfor details.
(Too) Few Assignments?
Possibly Err is much lower than the actual genotyping error rate.
Alternatively, a true parent will not be assigned when it is:
unclear who is the parent and who the offspring, due to unknown birth year for one or both individuals
unclear whether the parent is the father or mother
unclear if it is a parent or e.g. full sibling or grandparent, due to insufficient genetic data
And true half-siblings will not be clustered when it is:
unclear if they are maternal or paternal half-siblings
unclear if they are half-siblings, full avuncular, or grand-parental
unclear what type of relatives they are due to insufficient genetic data
All pairs of non-assigned but likely/definitely relatives can be found with
GetMaybeRel (run without conditioning on a pedigree). For a
method to do pairwise 'assignments', see
https://jiscah.github.io/articles/pairLL_classification.html ; for further
information, see the vignette.
If you already had a partial pedigree, running CalcOHLLR or
CalcParentProbs on it with the new SNP data and various
parameter value combinations may be informative.
Disclaimer
While every effort has been made to ensure that sequoia provides what it claims to do, there is absolutely no guarantee that the results provided are correct. Use of sequoia is entirely at your own risk.
References
Huisman, J. (2017) Pedigree reconstruction from SNP data: Parentage assignment, sibship clustering, and beyond. Molecular Ecology Resources 17:1009–1024.
See also
GenoConvertto read in various data formats,CheckGeno,SnpStatsto calculate missingness and allele frequencies,SimGenoto simulate SNP data from a pedigree,MakeAgePriorto estimate effect of age on relationships andPlotAgePriorto visualise those,SummarySeqandPlotPropAssignedto visualise results,GetMaybeRelto find pairs of potential relatives,GetRelMto turn a pedigree into pairwise relationships,CountOH,CalcPairLLandLLtoProbfor specified pairs of individuals respectively count Opposing Homozygous SNPs, calculate likelihoods of various relationships, and transform those likelihoods to probabilities,CalcOHLLRto count Opposing Homozygous SNPs and calculate LLR for all parent-offspring pairs in any pedigree,CalcParentProbsto calculate assignment probabilities (instead of LLRs) in any pedigree,CalcBYprobsto estimate birth years,PedCompareandComparePairsto compare two pedigrees,EstConfto estimate assignment errors,writeSeqto savesequoiaoutput as text or excel files,vignette("sequoia")for detailed manual & FAQ.
Author
Jisca Huisman, jisca.huisman@gmail.com
Examples
# === EXAMPLE 1: simulated data ===
head(SimGeno_example[,1:10])
#> V2 V3 V4 V5 V6 V7 V8 V9 V10 V11
#> a00013 0 0 0 1 0 0 0 1 0 2
#> a00008 1 1 1 1 2 1 1 1 1 0
#> a00011 0 2 1 2 2 1 0 2 0 0
#> a00023 0 0 1 1 1 0 0 0 2 0
#> a00006 1 1 1 0 0 0 0 0 1 1
#> a00004 0 1 1 1 2 1 1 0 0 1
head(LH_HSg5)
#> ID Sex BirthYear
#> 1 a00001 1 2000
#> 2 a00002 1 2000
#> 3 a00003 1 2000
#> 4 a00004 1 2000
#> 5 a00005 1 2000
#> 6 a00006 1 2000
# parentage assignment:
SeqOUT <- sequoia(GenoM = SimGeno_example, Err = 0.005,
LifeHistData = LH_HSg5, Module="par", Plot=TRUE)
#> ℹ Checking input data ...
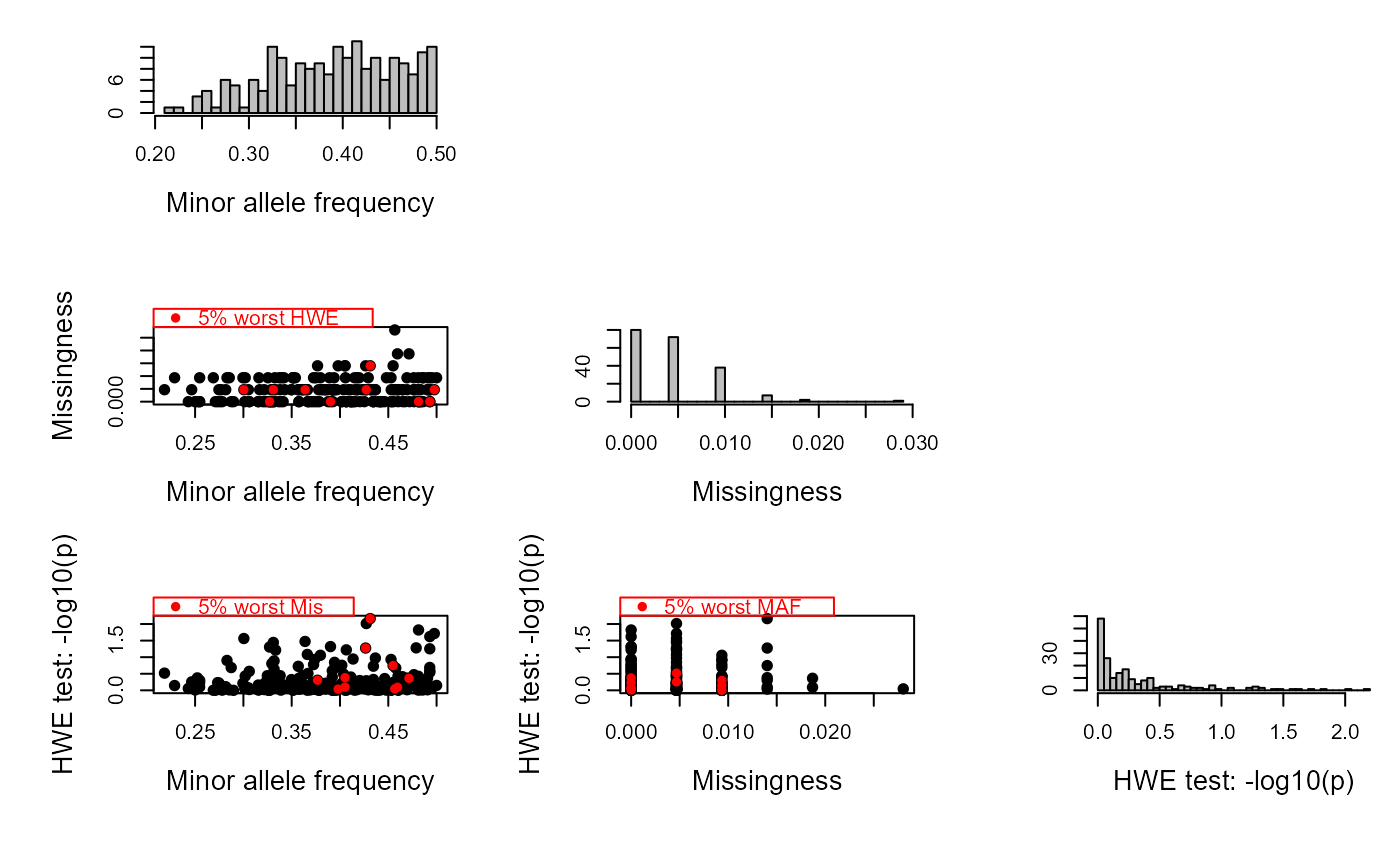 #> ✔ Genotype matrix looks OK! There are 214 individuals and 200 SNPs.
#>
#> ── Among genotyped individuals: ___
#> ℹ There are 106 females, 108 males, 0 of unknown sex, and 0 hermaphrodites.
#> ℹ Exact birth years are from 2000 to 2001
#> ___
#> ℹ Calling `MakeAgePrior()` ...
#> ℹ Ageprior: Flat 0/1, overlapping generations, MaxAgeParent = 2,2
#> ✔ Genotype matrix looks OK! There are 214 individuals and 200 SNPs.
#>
#> ── Among genotyped individuals: ___
#> ℹ There are 106 females, 108 males, 0 of unknown sex, and 0 hermaphrodites.
#> ℹ Exact birth years are from 2000 to 2001
#> ___
#> ℹ Calling `MakeAgePrior()` ...
#> ℹ Ageprior: Flat 0/1, overlapping generations, MaxAgeParent = 2,2
 #>
#> ~~~ Duplicate check ~~~
#> ✔ No potential duplicates found
#>
#> ~~~ Parentage assignment ~~~
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:56:58 | 0 | initial | | 0 | 0 | 0 | -18301.9
#> 15:56:58 | 0 | parents | | 130 | 167 | 0 | -13484.2
#>
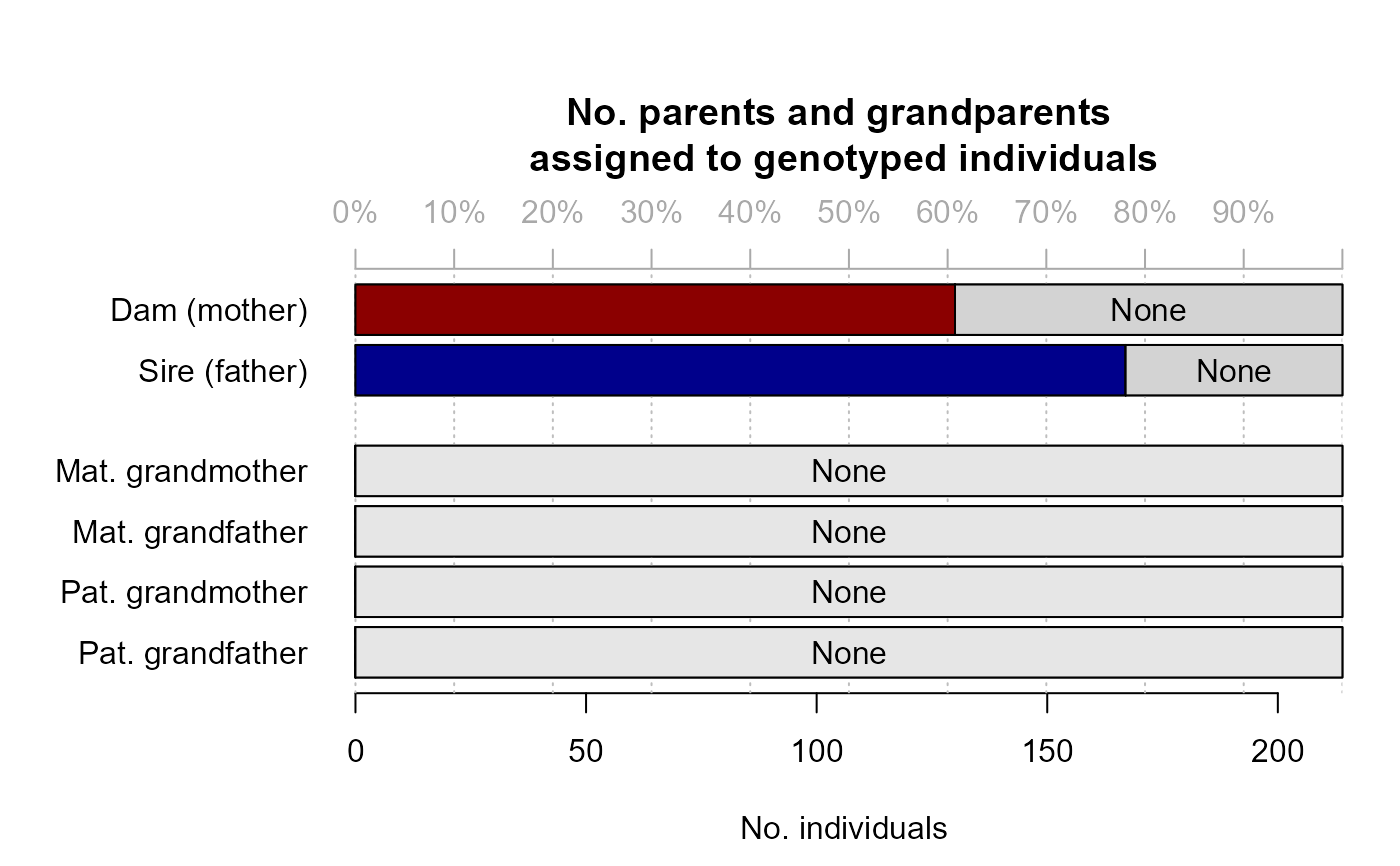
#> ✔ assigned 130 dams and 167 sires to 214 individuals
#>
#>
#> ~~~ Duplicate check ~~~
#> ✔ No potential duplicates found
#>
#> ~~~ Parentage assignment ~~~
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:56:58 | 0 | initial | | 0 | 0 | 0 | -18301.9
#> 15:56:58 | 0 | parents | | 130 | 167 | 0 | -13484.2
#>
#> ✔ assigned 130 dams and 167 sires to 214 individuals
#>
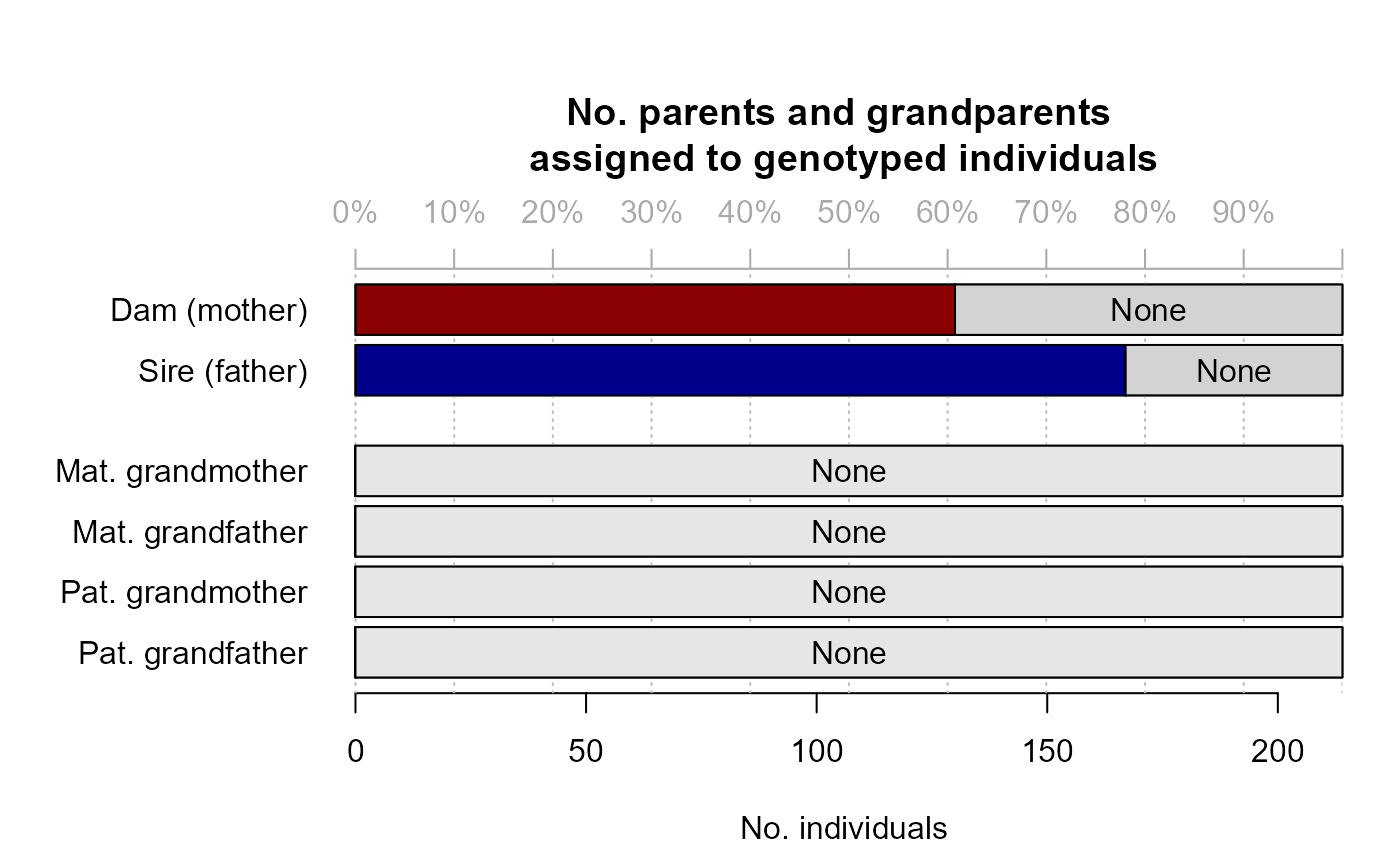 names(SeqOUT)
#> [1] "Specs" "ErrM" "args.AP" "DupLifeHistID"
#> [5] "NoLH" "AgePriors" "LifeHist" "PedigreePar"
#> [9] "TotLikPar" "LifeHistPar"
SeqOUT$PedigreePar[34:42, ]
#> id dam sire LLRdam LLRsire LLRpair OHdam OHsire MEpair
#> 34 a01002 <NA> <NA> NA NA NA NA NA NA
#> 35 b01003 <NA> <NA> NA NA NA NA NA NA
#> 36 b01004 <NA> <NA> NA NA NA NA NA NA
#> 37 a01005 a00013 b00001 NA NA NA 1 0 1
#> 38 b01006 a00013 b00001 NA NA NA 1 0 1
#> 39 b01007 a00013 b00001 NA NA NA 1 0 2
#> 40 a01008 a00013 b00001 NA NA NA 2 0 2
#> 41 b01009 a00008 b00016 NA NA NA 1 0 2
#> 42 a01010 a00008 b00016 NA NA NA 0 0 1
# compare to true (or old) pedigree:
PC <- PedCompare(Ped_HSg5, SeqOUT$PedigreePar)
names(SeqOUT)
#> [1] "Specs" "ErrM" "args.AP" "DupLifeHistID"
#> [5] "NoLH" "AgePriors" "LifeHist" "PedigreePar"
#> [9] "TotLikPar" "LifeHistPar"
SeqOUT$PedigreePar[34:42, ]
#> id dam sire LLRdam LLRsire LLRpair OHdam OHsire MEpair
#> 34 a01002 <NA> <NA> NA NA NA NA NA NA
#> 35 b01003 <NA> <NA> NA NA NA NA NA NA
#> 36 b01004 <NA> <NA> NA NA NA NA NA NA
#> 37 a01005 a00013 b00001 NA NA NA 1 0 1
#> 38 b01006 a00013 b00001 NA NA NA 1 0 1
#> 39 b01007 a00013 b00001 NA NA NA 1 0 2
#> 40 a01008 a00013 b00001 NA NA NA 2 0 2
#> 41 b01009 a00008 b00016 NA NA NA 1 0 2
#> 42 a01010 a00008 b00016 NA NA NA 0 0 1
# compare to true (or old) pedigree:
PC <- PedCompare(Ped_HSg5, SeqOUT$PedigreePar)
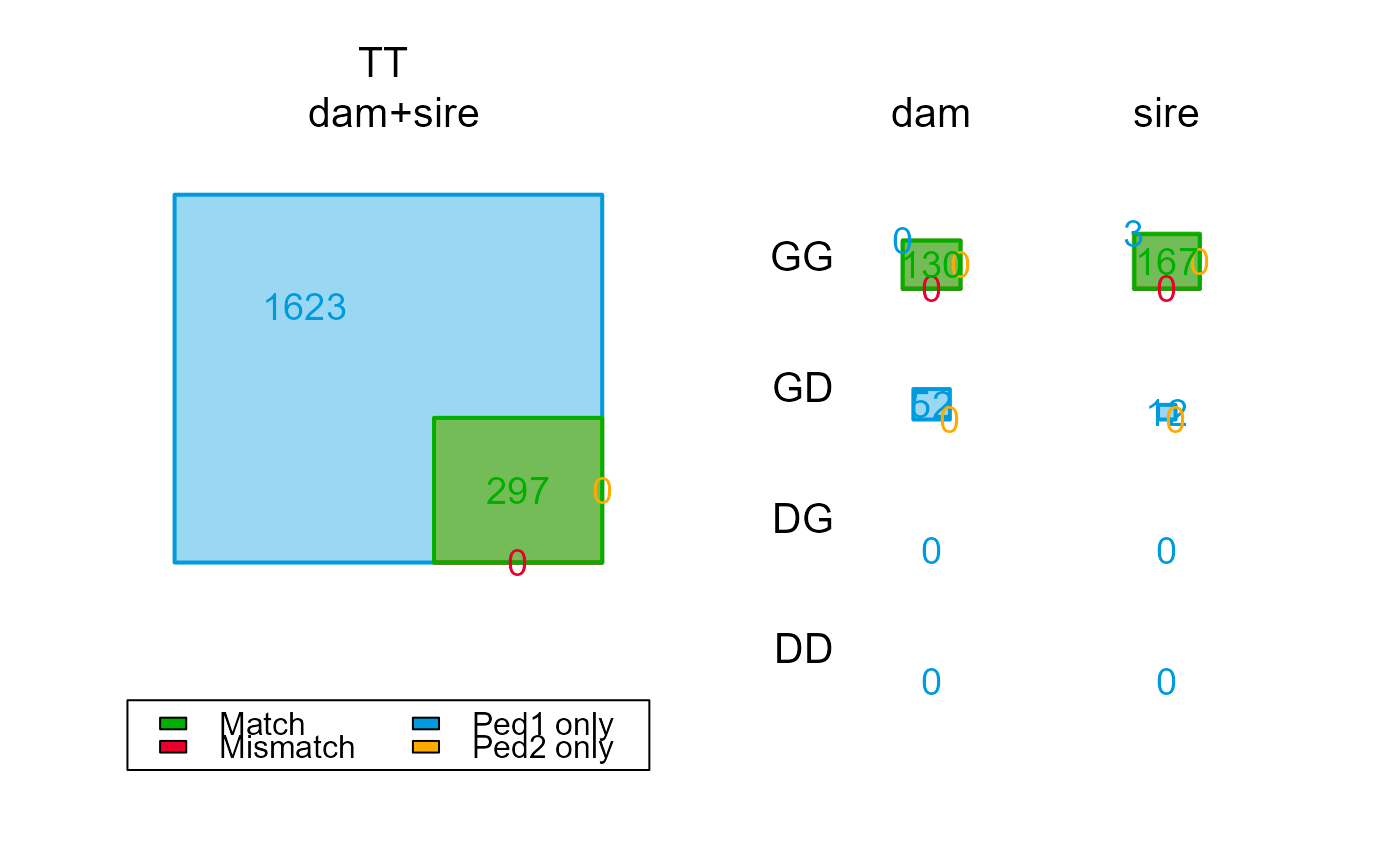 PC$Counts["GG",,]
#> parent
#> class dam sire
#> Total 130 170
#> Match 130 167
#> Mismatch 0 0
#> P1only 0 3
#> P2only 0 0
# \donttest{
# parentage assignment + full pedigree reconstruction:
# (note: this can be rather time consuming)
SeqOUT2 <- sequoia(GenoM = SimGeno_example, Err = 0.005,
LifeHistData = LH_HSg5, Module="ped", quiet="verbose")
#> ℹ Checking input data ...
PC$Counts["GG",,]
#> parent
#> class dam sire
#> Total 130 170
#> Match 130 167
#> Mismatch 0 0
#> P1only 0 3
#> P2only 0 0
# \donttest{
# parentage assignment + full pedigree reconstruction:
# (note: this can be rather time consuming)
SeqOUT2 <- sequoia(GenoM = SimGeno_example, Err = 0.005,
LifeHistData = LH_HSg5, Module="ped", quiet="verbose")
#> ℹ Checking input data ...
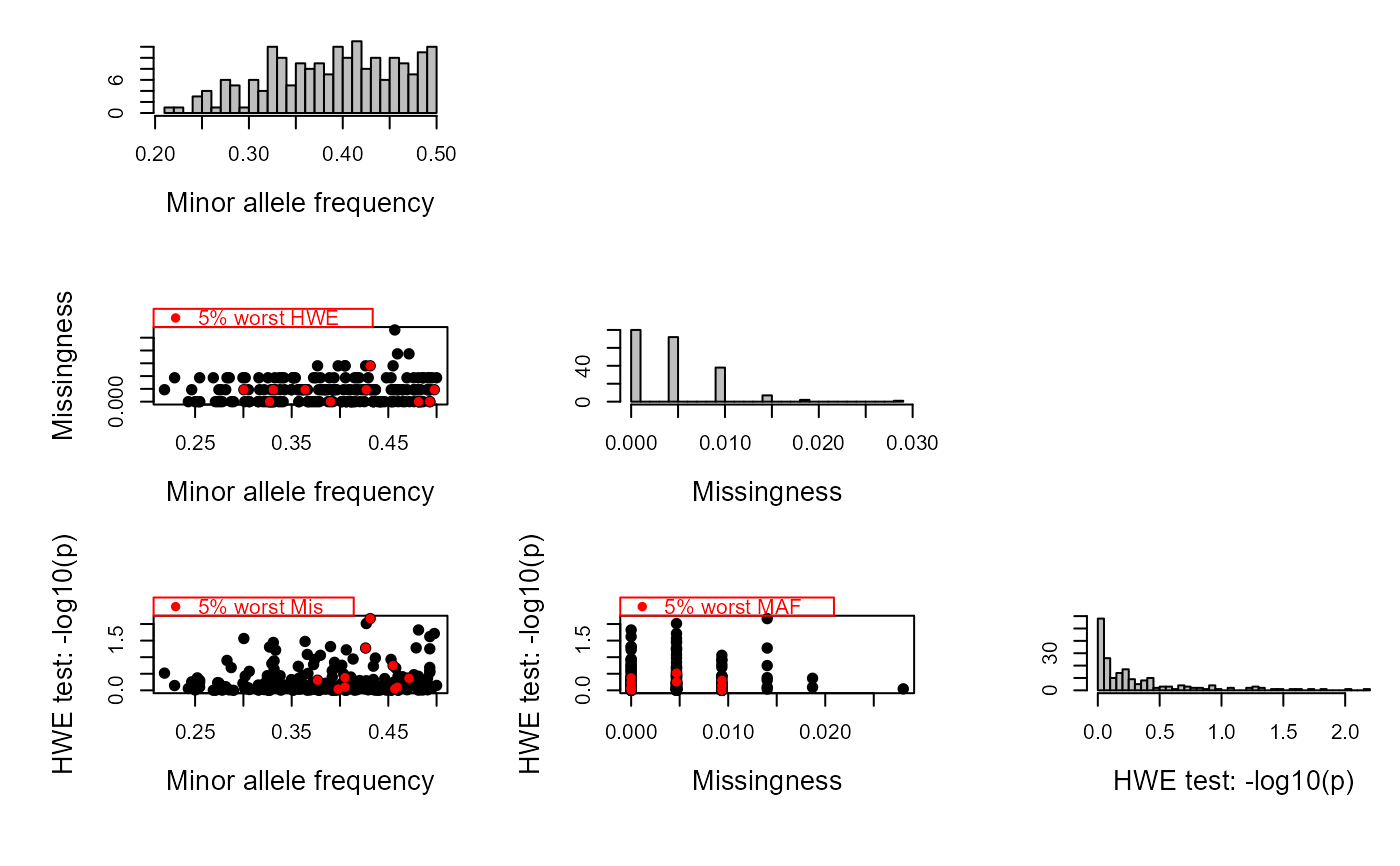 #> ✔ Genotype matrix looks OK! There are 214 individuals and 200 SNPs.
#>
#> ── Among genotyped individuals: ___
#> ℹ There are 106 females, 108 males, 0 of unknown sex, and 0 hermaphrodites.
#> ℹ Exact birth years are from 2000 to 2001
#> ___
#> ℹ Calling `MakeAgePrior()` ...
#> ℹ Ageprior: Flat 0/1, overlapping generations, MaxAgeParent = 2,2
#> ✔ Genotype matrix looks OK! There are 214 individuals and 200 SNPs.
#>
#> ── Among genotyped individuals: ___
#> ℹ There are 106 females, 108 males, 0 of unknown sex, and 0 hermaphrodites.
#> ℹ Exact birth years are from 2000 to 2001
#> ___
#> ℹ Calling `MakeAgePrior()` ...
#> ℹ Ageprior: Flat 0/1, overlapping generations, MaxAgeParent = 2,2
 #>
#> ~~~ Duplicate check ~~~
#> 0 10 20 30 40 50 60 70 80 90 100%
#> | | | | | | | | | | |
#> ****************************************
#> ✔ No potential duplicates found
#>
#> ~~~ Parentage assignment ~~~
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:01 | 0 | count OH | |
#> 15:57:01 | 0 | initial | | 0 | 0 | 0 | -18301.9
#> 15:57:01 | 1 | parents | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:02 | 2 | parents | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:02 | 99 | est byears | .......... |
#>
#> ✔ assigned 130 dams and 167 sires to 214 individuals
#>
#>
#> ~~~ Duplicate check ~~~
#> 0 10 20 30 40 50 60 70 80 90 100%
#> | | | | | | | | | | |
#> ****************************************
#> ✔ No potential duplicates found
#>
#> ~~~ Parentage assignment ~~~
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:01 | 0 | count OH | |
#> 15:57:01 | 0 | initial | | 0 | 0 | 0 | -18301.9
#> 15:57:01 | 1 | parents | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:02 | 2 | parents | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:02 | 99 | est byears | .......... |
#>
#> ✔ assigned 130 dams and 167 sires to 214 individuals
#>
 #> ℹ Ageprior: Flat 0/1, discrete generations, MaxAgeParent = 1,1
#> ℹ Ageprior: Flat 0/1, discrete generations, MaxAgeParent = 1,1
 #>
#> ~~~ Full pedigree reconstruction ~~~
#> Transferring input pedigree ...
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:03 | 0 | count OH | |
#> 15:57:03 | 0 | initial | | 130 | 167 | 0 | -13484.2
#> 15:57:03 | 0 | ped check | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:03 | 1 | find pairs | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:03 | 1 | clustering | .......... | 181 | 181 | 0 | -12354.3
#> 15:57:03 | 1 | merging | .......... | 181 | 181 | 0 | -12354.3
#> 15:57:03 | 1 | P of sibs | .......... | 181 | 181 | 0 | -12354.3
#> 15:57:04 | 1 | find/check | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | find pairs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | clustering | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | merging | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | P of sibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | GP Hsibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | find/check | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | find pairs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | clustering | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | GP pairs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | merging | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | P of sibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | GP Hsibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | GP Fsibs | | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | find/check | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 99 | est byears | .......... |
#>
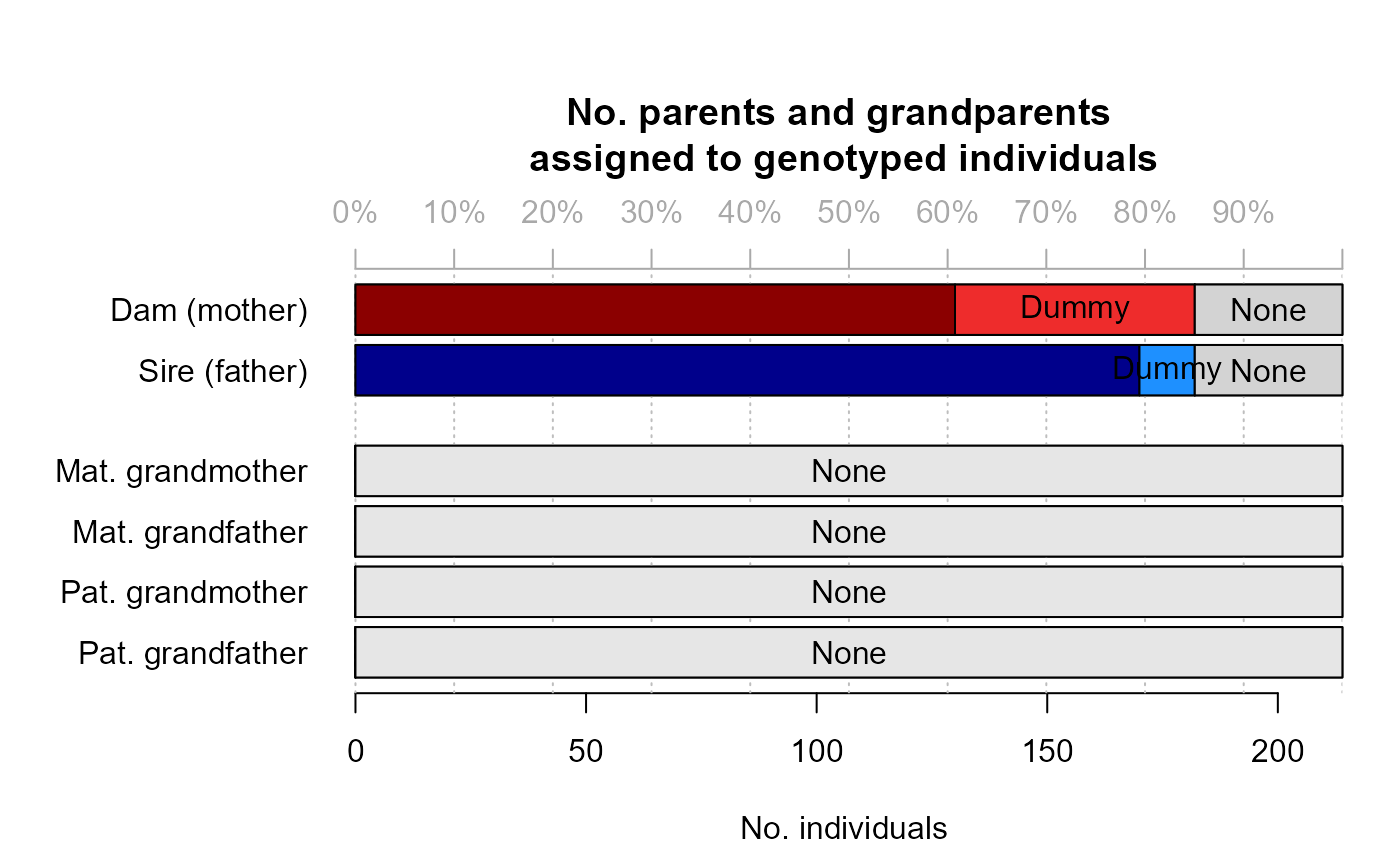
#> ✔ assigned 182 dams and 182 sires to 214 + 8 individuals (real + dummy)
#>
#> ℹ You can use `SummarySeq()` for pedigree details, `CalcParentProbs()` for
#> assignment probabilities, and `EstConf()` for confidence estimates
#> ℹ Run `GetMaybeRel()` conditional on this pedigree to check for any
#> non-assigned relatives
#>
#> ~~~ Full pedigree reconstruction ~~~
#> Transferring input pedigree ...
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:03 | 0 | count OH | |
#> 15:57:03 | 0 | initial | | 130 | 167 | 0 | -13484.2
#> 15:57:03 | 0 | ped check | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:03 | 1 | find pairs | .......... | 130 | 167 | 0 | -13484.2
#> 15:57:03 | 1 | clustering | .......... | 181 | 181 | 0 | -12354.3
#> 15:57:03 | 1 | merging | .......... | 181 | 181 | 0 | -12354.3
#> 15:57:03 | 1 | P of sibs | .......... | 181 | 181 | 0 | -12354.3
#> 15:57:04 | 1 | find/check | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | find pairs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | clustering | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | merging | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | P of sibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | GP Hsibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 2 | find/check | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | find pairs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | clustering | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | GP pairs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | merging | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | P of sibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | GP Hsibs | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | GP Fsibs | | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 3 | find/check | .......... | 182 | 182 | 0 | -12315.8
#> 15:57:04 | 99 | est byears | .......... |
#>
#> ✔ assigned 182 dams and 182 sires to 214 + 8 individuals (real + dummy)
#>
#> ℹ You can use `SummarySeq()` for pedigree details, `CalcParentProbs()` for
#> assignment probabilities, and `EstConf()` for confidence estimates
#> ℹ Run `GetMaybeRel()` conditional on this pedigree to check for any
#> non-assigned relatives
 SeqOUT2$Pedigree[34:42, ]
#> id dam sire LLRdam LLRsire LLRpair OHdam OHsire MEpair
#> 34 a01002 F0005 M0001 NA NA NA NA NA NA
#> 35 b01003 F0005 M0001 NA NA NA NA NA NA
#> 36 b01004 F0005 M0001 NA NA NA NA NA NA
#> 37 a01005 a00013 b00001 NA NA NA 1 0 1
#> 38 b01006 a00013 b00001 NA NA NA 1 0 1
#> 39 b01007 a00013 b00001 NA NA NA 1 0 2
#> 40 a01008 a00013 b00001 NA NA NA 2 0 2
#> 41 b01009 a00008 b00016 NA NA NA 1 0 2
#> 42 a01010 a00008 b00016 NA NA NA 0 0 1
PC2 <- PedCompare(Ped_HSg5, SeqOUT2$Pedigree)
SeqOUT2$Pedigree[34:42, ]
#> id dam sire LLRdam LLRsire LLRpair OHdam OHsire MEpair
#> 34 a01002 F0005 M0001 NA NA NA NA NA NA
#> 35 b01003 F0005 M0001 NA NA NA NA NA NA
#> 36 b01004 F0005 M0001 NA NA NA NA NA NA
#> 37 a01005 a00013 b00001 NA NA NA 1 0 1
#> 38 b01006 a00013 b00001 NA NA NA 1 0 1
#> 39 b01007 a00013 b00001 NA NA NA 1 0 2
#> 40 a01008 a00013 b00001 NA NA NA 2 0 2
#> 41 b01009 a00008 b00016 NA NA NA 1 0 2
#> 42 a01010 a00008 b00016 NA NA NA 0 0 1
PC2 <- PedCompare(Ped_HSg5, SeqOUT2$Pedigree)
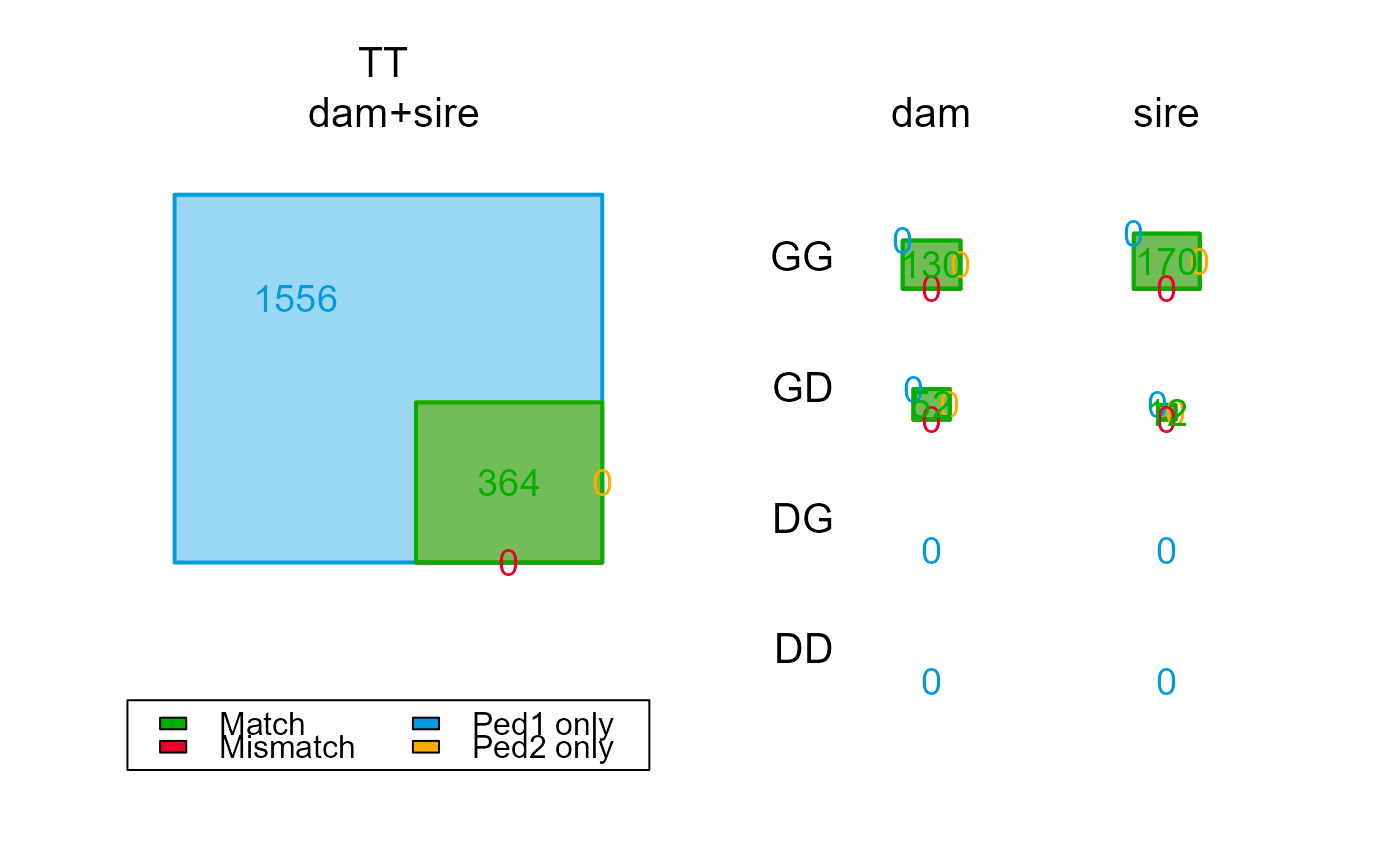 PC2$Counts["GT",,]
#> parent
#> class dam sire
#> Total 182 182
#> Match 182 182
#> Mismatch 0 0
#> P1only 0 0
#> P2only 0 0
PC2$Counts[,,"dam"]
#> class
#> cat Total Match Mismatch P1only P2only
#> GG 130 130 0 0 0
#> GD 52 52 0 0 0
#> GT 182 182 0 0 0
#> DG 0 0 0 0 0
#> DD 0 0 0 0 0
#> DT 0 0 0 0 0
#> TT 960 182 0 778 0
# different kind of pedigree comparison:
ComparePairs(Ped1=Ped_HSg5, Ped2=SeqOUT$PedigreePar, patmat=TRUE)
#> Ped2
#> Ped1 M P O FS MHS PHS U X
#> M 130 0 0 0 0 0 0 830
#> P 0 167 0 0 0 0 3 790
#> FS 0 0 0 206 12 59 13 1310
#> MHS 0 0 0 0 217 0 97 1446
#> PHS 0 0 0 0 0 587 71 3022
#> U 0 0 0 0 0 0 21229 469311
#> X 0 0 0 0 0 0 0 0
# results overview:
SummarySeq(SeqOUT2)
PC2$Counts["GT",,]
#> parent
#> class dam sire
#> Total 182 182
#> Match 182 182
#> Mismatch 0 0
#> P1only 0 0
#> P2only 0 0
PC2$Counts[,,"dam"]
#> class
#> cat Total Match Mismatch P1only P2only
#> GG 130 130 0 0 0
#> GD 52 52 0 0 0
#> GT 182 182 0 0 0
#> DG 0 0 0 0 0
#> DD 0 0 0 0 0
#> DT 0 0 0 0 0
#> TT 960 182 0 778 0
# different kind of pedigree comparison:
ComparePairs(Ped1=Ped_HSg5, Ped2=SeqOUT$PedigreePar, patmat=TRUE)
#> Ped2
#> Ped1 M P O FS MHS PHS U X
#> M 130 0 0 0 0 0 0 830
#> P 0 167 0 0 0 0 3 790
#> FS 0 0 0 206 12 59 13 1310
#> MHS 0 0 0 0 217 0 97 1446
#> PHS 0 0 0 0 0 587 71 3022
#> U 0 0 0 0 0 0 21229 469311
#> X 0 0 0 0 0 0 0 0
# results overview:
SummarySeq(SeqOUT2)
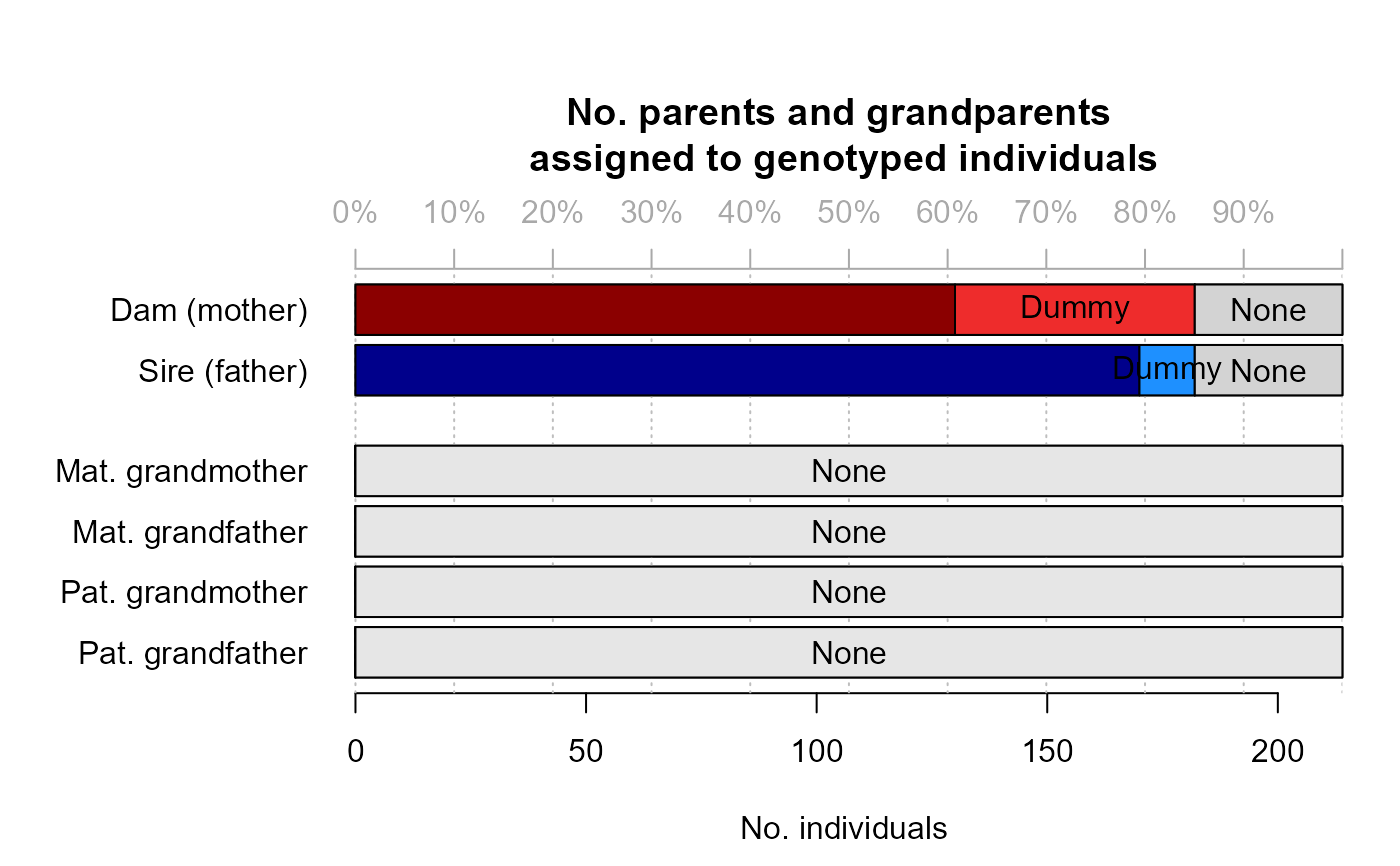
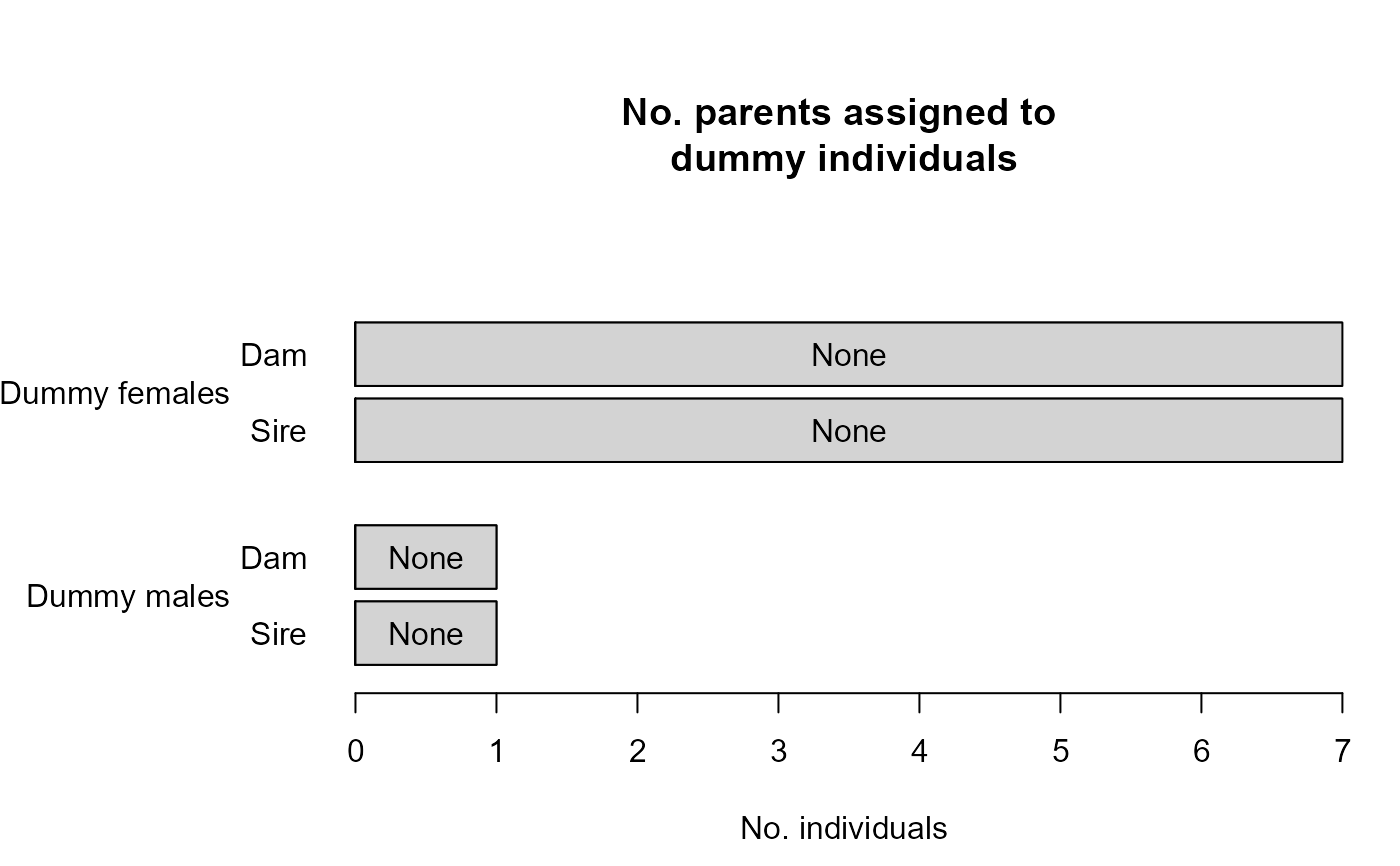
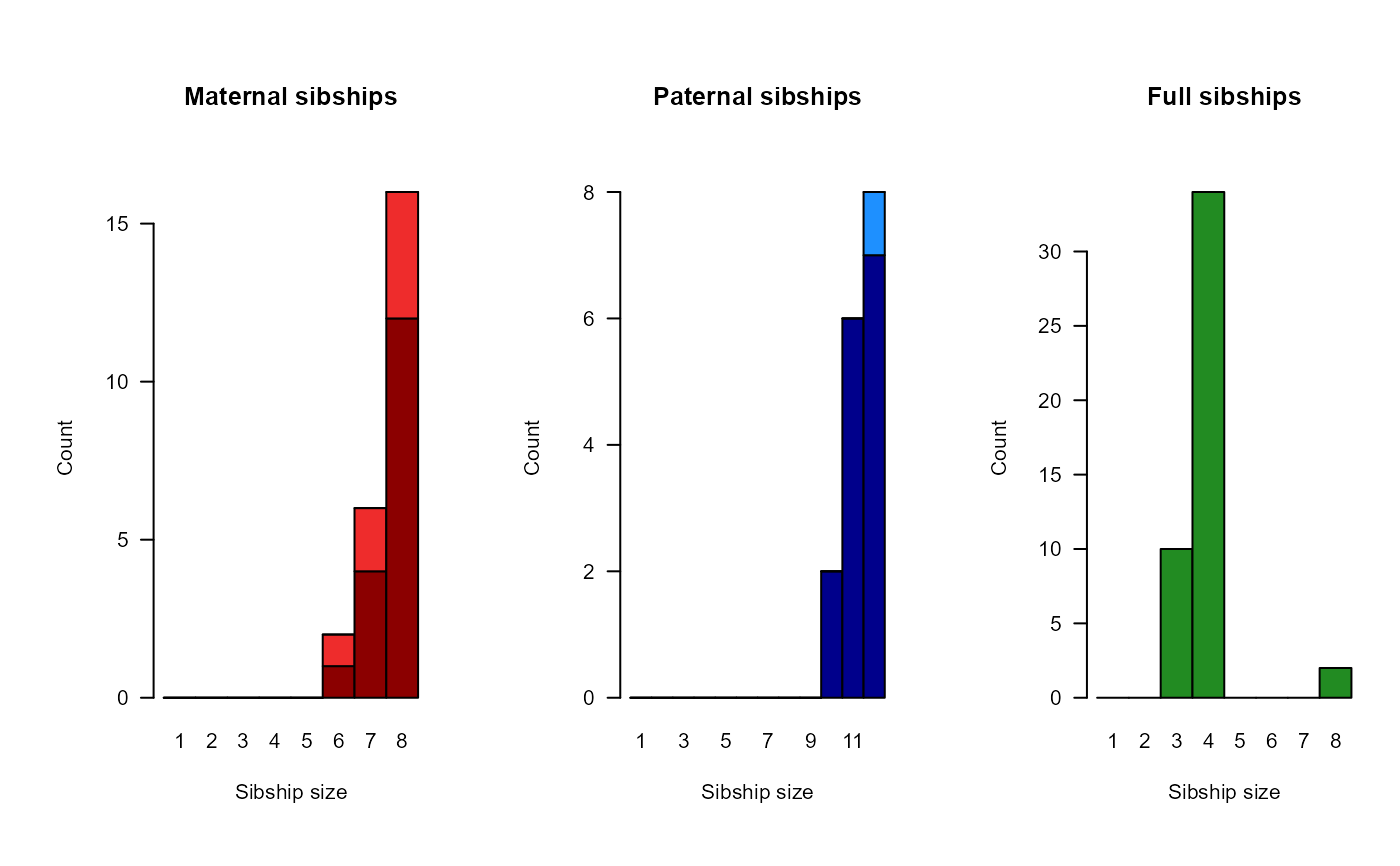
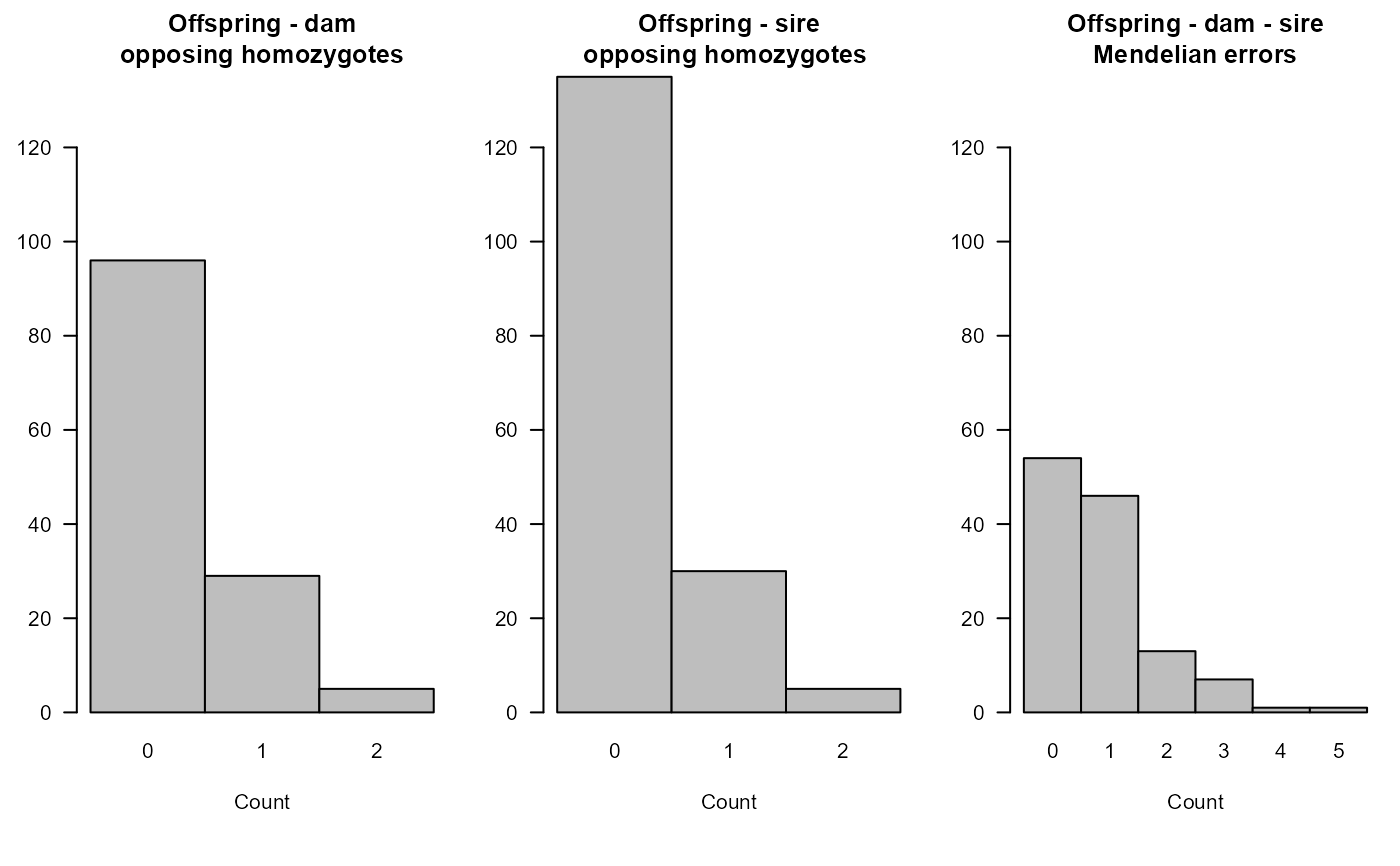 #> ℹ No `LLR` panel, because LLR columns are all `<NA>`
# important to run with approx. correct genotyping error rate:
SeqOUT2.b <- sequoia(GenoM = SimGeno_example, # Err = 1e-4 by default
LifeHistData = LH_HSg5, Module="ped", Plot=FALSE)
#> ℹ Checking input data ...
#> ✔ Genotype matrix looks OK! There are 214 individuals and 200 SNPs.
#>
#> ── Among genotyped individuals: ___
#> ℹ There are 106 females, 108 males, 0 of unknown sex, and 0 hermaphrodites.
#> ℹ Exact birth years are from 2000 to 2001
#> ___
#> ℹ Calling `MakeAgePrior()` ...
#> ℹ Ageprior: Flat 0/1, overlapping generations, MaxAgeParent = 2,2
#>
#> ~~~ Duplicate check ~~~
#> ✔ No potential duplicates found
#>
#> ~~~ Parentage assignment ~~~
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:05 | 0 | initial | | 0 | 0 | 0 | -18301.9
#> 15:57:05 | 0 | parents | | 125 | 162 | 0 | -13732.7
#>
#> ✔ assigned 125 dams and 162 sires to 214 individuals
#>
#> ℹ Ageprior: Flat 0/1, discrete generations, MaxAgeParent = 1,1
#>
#> ~~~ Full pedigree reconstruction ~~~
#> Transferring input pedigree ...
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:06 | 0 | initial | | 125 | 162 | 0 | -13732.7
#> 15:57:07 | 1 | (all) | | 180 | 180 | 0 | -12512.9
#> 15:57:08 | 2 | (all) | | 180 | 180 | 0 | -12512.9
#> 15:57:08 | 3 | (all) | | 180 | 180 | 0 | -12512.9
#>
#> ✔ assigned 180 dams and 180 sires to 214 + 9 individuals (real + dummy)
#>
PC2.b <- PedCompare(Ped_HSg5, SeqOUT2.b$Pedigree)
#> ℹ No `LLR` panel, because LLR columns are all `<NA>`
# important to run with approx. correct genotyping error rate:
SeqOUT2.b <- sequoia(GenoM = SimGeno_example, # Err = 1e-4 by default
LifeHistData = LH_HSg5, Module="ped", Plot=FALSE)
#> ℹ Checking input data ...
#> ✔ Genotype matrix looks OK! There are 214 individuals and 200 SNPs.
#>
#> ── Among genotyped individuals: ___
#> ℹ There are 106 females, 108 males, 0 of unknown sex, and 0 hermaphrodites.
#> ℹ Exact birth years are from 2000 to 2001
#> ___
#> ℹ Calling `MakeAgePrior()` ...
#> ℹ Ageprior: Flat 0/1, overlapping generations, MaxAgeParent = 2,2
#>
#> ~~~ Duplicate check ~~~
#> ✔ No potential duplicates found
#>
#> ~~~ Parentage assignment ~~~
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:05 | 0 | initial | | 0 | 0 | 0 | -18301.9
#> 15:57:05 | 0 | parents | | 125 | 162 | 0 | -13732.7
#>
#> ✔ assigned 125 dams and 162 sires to 214 individuals
#>
#> ℹ Ageprior: Flat 0/1, discrete generations, MaxAgeParent = 1,1
#>
#> ~~~ Full pedigree reconstruction ~~~
#> Transferring input pedigree ...
#>
#> Time | R | Step | Progress | Dams | Sires | GPs | Total LL
#> -------- | -- | ---------- | ---------- | ----- | ----- | ----- | ----------
#> 15:57:06 | 0 | initial | | 125 | 162 | 0 | -13732.7
#> 15:57:07 | 1 | (all) | | 180 | 180 | 0 | -12512.9
#> 15:57:08 | 2 | (all) | | 180 | 180 | 0 | -12512.9
#> 15:57:08 | 3 | (all) | | 180 | 180 | 0 | -12512.9
#>
#> ✔ assigned 180 dams and 180 sires to 214 + 9 individuals (real + dummy)
#>
PC2.b <- PedCompare(Ped_HSg5, SeqOUT2.b$Pedigree)
 PC2.b$Counts["GT",,]
#> parent
#> class dam sire
#> Total 182 182
#> Match 178 180
#> Mismatch 2 0
#> P1only 2 2
#> P2only 0 0
# }
if (FALSE) { # \dontrun{
# === EXAMPLE 2: real data ===
# ideally, select 400-700 SNPs: high MAF & low LD
# save in 0/1/2/NA format (PLINK's --recodeA)
GenoM <- GenoConvert(InFile = "inputfile_for_sequoia.raw",
InFormat = "raw") # can also do Colony format
SNPSTATS <- SnpStats(GenoM)
# perhaps after some data-cleaning:
write.table(GenoM, file="MyGenoData.txt", row.names=T, col.names=F)
# later:
GenoM <- as.matrix(read.table("MyGenoData.txt", row.names=1, header=FALSE))
# or for very large datasets:
GenoM <- data.table::fread('MyGenoData.txt') %>% as.matrix(rownames=1)
LHdata <- read.table("LifeHistoryData.txt", header=T) # ID-Sex-birthyear
SeqOUT <- sequoia(GenoM, LHdata, Err=0.005)
SummarySeq(SeqOUT)
SeqOUT$notes <- "Trial run on cleaned data" # add notes for future reference
saveRDS(SeqOUT, file="sequoia_output_42.RDS") # save to R-specific file
writeSeq(SeqOUT, folder="sequoia_output") # save to several plain text files
# runtime:
SeqOUT$Specs$TimeEnd - SeqOUT$Specs$TimeStart
} # }
PC2.b$Counts["GT",,]
#> parent
#> class dam sire
#> Total 182 182
#> Match 178 180
#> Mismatch 2 0
#> P1only 2 2
#> P2only 0 0
# }
if (FALSE) { # \dontrun{
# === EXAMPLE 2: real data ===
# ideally, select 400-700 SNPs: high MAF & low LD
# save in 0/1/2/NA format (PLINK's --recodeA)
GenoM <- GenoConvert(InFile = "inputfile_for_sequoia.raw",
InFormat = "raw") # can also do Colony format
SNPSTATS <- SnpStats(GenoM)
# perhaps after some data-cleaning:
write.table(GenoM, file="MyGenoData.txt", row.names=T, col.names=F)
# later:
GenoM <- as.matrix(read.table("MyGenoData.txt", row.names=1, header=FALSE))
# or for very large datasets:
GenoM <- data.table::fread('MyGenoData.txt') %>% as.matrix(rownames=1)
LHdata <- read.table("LifeHistoryData.txt", header=T) # ID-Sex-birthyear
SeqOUT <- sequoia(GenoM, LHdata, Err=0.005)
SummarySeq(SeqOUT)
SeqOUT$notes <- "Trial run on cleaned data" # add notes for future reference
saveRDS(SeqOUT, file="sequoia_output_42.RDS") # save to R-specific file
writeSeq(SeqOUT, folder="sequoia_output") # save to several plain text files
# runtime:
SeqOUT$Specs$TimeEnd - SeqOUT$Specs$TimeStart
} # }