SNP Summary Statistics
SnpStats.RdEstimate allele frequency (AF), missingness and Mendelian errors per SNP.
Usage
SnpStats(
GenoM,
Pedigree = NULL,
Duplicates = NULL,
Plot = TRUE,
quiet = TRUE,
calc_HWE = TRUE,
ErrFlavour
)Arguments
- GenoM
genotype matrix, in sequoia's format: 1 column per SNP, 1 row per individual, genotypes coded as 0/1/2/-9, and row names giving individual IDs.
- Pedigree
dataframe with 3 columns: ID - parent1 - parent2. Additional columns and non-genotyped individuals are ignored. Used to count Mendelian errors per SNP and (poorly) estimate the error rate.
- Duplicates
dataframe with pairs of duplicated samples
- Plot
logical, show histograms of the results?
- quiet
logical, suppress messages?
- calc_HWE
logical, calculate chi-square test for Hardy-Weinberg equilibrium? Can be relatively time consuming for large datasets.
- ErrFlavour
DEPRECATED AND IGNORED. Was used to estimate
Err.hat
Value
A matrix with a number of rows equal to the number of SNPs (=number of columns of GenoM), and when no Pedigree is provided 2 columns:
- AF
Allele frequency of the 'second allele' (the one for which the homozygote is coded 2)
- Mis
Proportion of missing calls
- HWE.p
p-value from chi-square test for Hardy-Weinberg equilibrium
When a Pedigree is provided, there are 8 additional columns:
- n.dam, n.sire, n.pair
Number of dams, sires, parent-pairs successfully genotyped for the SNP
- OHdam, OHsire
Count of number of opposing homozygous cases
- MEpair
Count of Mendelian errors, includes opposing homozygous cases when only one parent is genotyped
- n.dups, n.diff
Number of duplicate pairs successfully genotyped for the SNP; number of differences. The latter does not count cases where one duplicate is not successfully genotyped at the SNP
Details
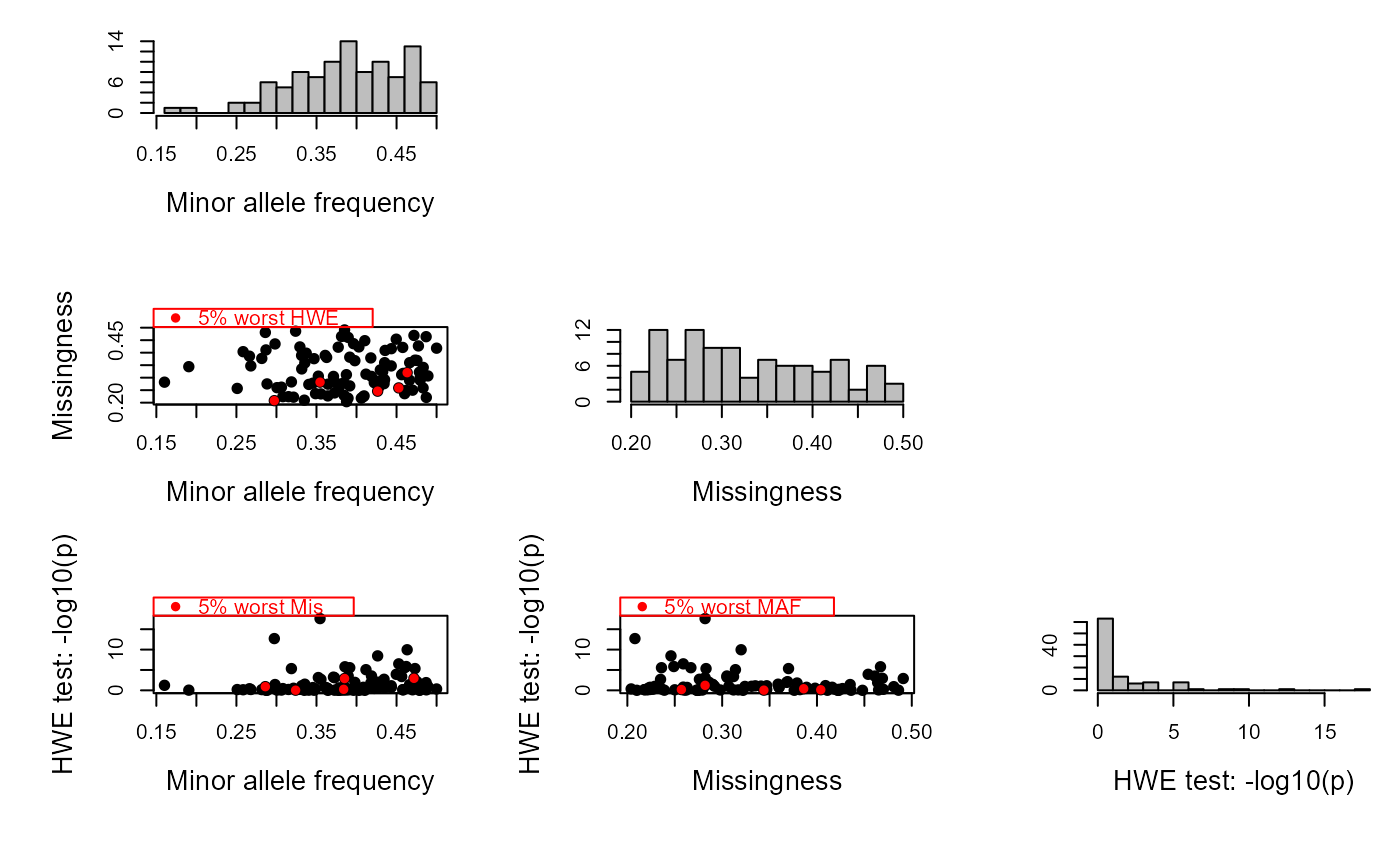
Calculation of these summary statistics can be done in PLINK, and SNPs with low minor allele frequency or high missingness should be filtered out prior to pedigree reconstruction. This function is provided as an aid to inspect the relationship between AF, missingness and genotyping error to find a suitable combination of SNP filtering thresholds to use.
For pedigree reconstruction, SNPs with zero or one copies of the alternate allele in the dataset (MAF \(\le 1/2N\)) are considered fixed, and excluded.
See also
GenoConvert to convert from various data formats;
CheckGeno to check the data is in valid format for sequoia
and exclude monomorphic SNPs etc., CalcOHLLR to calculate OH
& ME per individual.